Syndrome hémorragique d'origine hématologique : saignement anormal, induit ou majoré par une pathologie de l'hémostase, acquise ou constitutionnelle.
Physiologe de l'hémostase
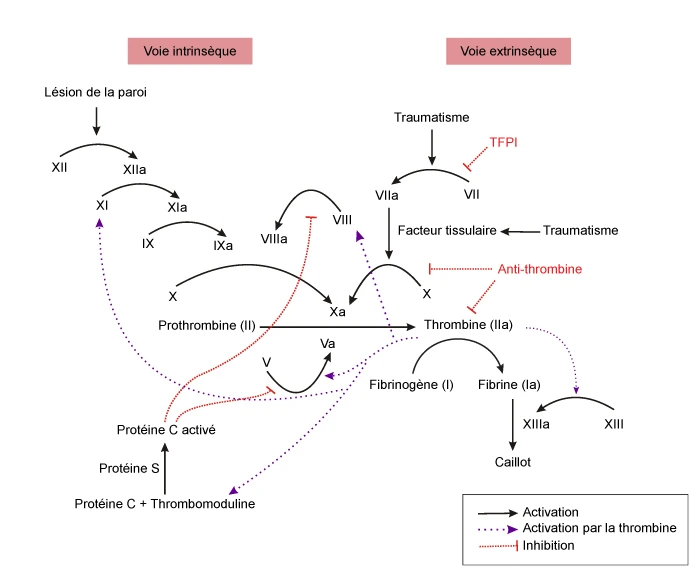
3 temps essentiels, étroitement imbriqués, durant lesquels participent cellules, protéines et phospholipides :
- Hémostase primaire : formation d'un thrombus à prédominance de plaquettes, avec la participation des cellules endothéliales, du facteur de von Willebrand (vWF) et du fibrinogène.
- Coagulation (hémostase secondaire) : la cascade de la coagulation, grâce à ses différents facteurs (dont la plupart sont synthétisés par le foie, et certains en présence de vitamine K), permet de former un caillot hémostatique solide.
- Fibrinolyse : le caillot est lysé par la plasmine, une enzyme générée après activation du plasminogène par le t-PA ou l'u-Pa (urokinase), ce qui entraîne la libération de produits de dégradation de la fibrine (PDF) et de D-Dimères.
Facteurs vitamine-K dépendants : II, VII, IX, X.
Orientation dès l'examen clinique
Les éléments de l'interrogatoire qui doivent faire suspecter une maladie hémorragique d'origine hématologique sont :
- Antécédents hémorragiques personnels : date de début des signes, types de saignement (cutané, muqueux, viscéral, articulaire, etc.), spontanés ou provoqués, après gestes invasifs / chirurgies, anémie, traitement martial, nécessité de transfusion, etc.
- Si femme : ménométrorragie, etc.
- Traitements médicamenteux : antiagrégants plaquettaires, antithrombotiques, etc.
- Antécédents hémorragiques familiaux : maladie de Willebrand, hémophilie, etc.
A l'examen physique, il faut rechercher :
- des saignements cutanés (purpura pétéchial, ecchymoses), muqueux (bouche, pharynx), profonds (hématome) ou articulaires (hémarthrose)
- des signes d'anémie, de carence martiale
- des pathologies sous-jacentes : insuffisance hépatique, rénale, infection, maladie auto-immune (lupus érythémateux disséminés surtout), hémopathie malagnie, cancer, etc.
Il est possible de s'orienter entre une atteinte de l'hémostase primaire et une atteinte de la coagulation :
- Hémostase primaire : hémorragies cutanées et muqueuses (purpura pétéchial et/ou ecchymotiques), saignements spontanées et/ou provoqués
- Coagulation : hémarthroses, hématomes profonds, saignement provoqué par un traumatisme minime, saignement retardé
Examens complémentaires
En première intention :
- Hémogramme
- Temps de Quick (ou taux de prothrombine, TP) : voie extrinsèque et voie commune
- Temps de céphaline avec activateur (TCA) : voie extrinsèque et voie commune
- Dosage du fibrinogène
A titre indicatif, voici les valeurs anormales et les limites entraînant un risque hémorragique :
| Anormal | Risque hémorragique | |
|---|---|---|
| Plaquettes (G/L) | < 150 | < 50 - 70 |
| Taux de prothrombine (%) | < 70 | < 30 |
| TCAr (rapport patient/témoin) | > 1,2 (ou 1,3 chez l'enfant) | > 1,2 (ou 1,3 chez l'enfant) |
| Fibrinogène (g/L) | < 2 | < 0,5 - 0,8 |
| Facteurs II, V, VII, IX, X, XI (%) | < 70 | < 30 |
| Facteur de Willebard et facteur VIII (%) | < 50 | < 30 |
En cas de thrombopénie : contrôler une fausse thrombopénie en vérifiant l'absence d'agrégats in vitro sur tube citraté ou hépariné, car possible agrégation dans les tubes de prélévement à cause de la présence d'acide éthylène diamine tétra-acétique (EDTA).
Si autres anomalies : examens plus spécifiques selon l'orientation (dosages des facteurs de coagulation, étude du facteur de Willebrand, analyse des fonctions plaquettaires, etc.)
Les déficits en facteurs de coagulation
1. Hémophilie constitutionnelle
- Clinique : garçons dès l'âge de la marche
- Test de dépistage le plus sensible : TCAr allongé
- Diagnostic : dosage des taux de facteurs VIII (hémophilie A) et IX (hémophilie B)
2. Déficits acquis de la coagulation
- Hypovitaminose K : TP diminué, facteur V normal
- Insuffisance hépatocellulaire : TP abaissé, facteur V abaissé
3. Coagulation intravasculaire disséminée (CIVD)
CIVD : conséquence de l'activation pathologique et diffuse de la coagulation.
Causes : multiples
- médicales : infections sévères, cancers, leucémies (LAM3 surtout), hémolyses intravasculaires, etc.
- obstétricales : hématome rétroplacentaire, embolie amniotique, toxémie gravidique, éclampsie, mort foetale in utero, môle hydatiforme, placenta praevia
- chirurgicales et traumatiques : chirurgie pulmonaire ou cardiaque avec circulation extracorporelle, prostatique, polytraumatismes, brûlures étendues, etc.
- autres : morsures de serpents, embolies graisseuses, malformations vasculaires, etc.
Manifestations : soit hémorragiques, soit thrombotiques
Anomalies :
- Les plus précoces : thrombopénie, fibrinogène abaissé
- Puis consommation variable des facteurs de coagulation (surtout le facteur V) : allongement du TCA, baisse du TP
- Enfin : augmentation des produits de dégradation de la fibrine (PDF) et des D-dimères
4. Hémophilie acquise
- Contexte : sujet âgé, jeune femme en post-partum
- A évoquer si : syndrome hémorragique récent, TCAr allongé et non corrigé par adition de plasma normal, avec taux de facteur VIII abaissé
Thrombopathie
Thrombopathie : maladie fonctionnelle des plaquettes.
A évoquer si : saignements cutanéomuqueux inexpliqués associés à des examens de débrouillage normaux.
Causes :
- acquises surtout : médicaments (antiagrégants plaquettaires, anti-inflammatoire, antibiotiques, antidépresseurs, etc.) et hémopathies (syndromes myéloprolifératifs ou myélodysplasiques, gammapathies monoclonales, etc.).
- constitutionnelles : thrombasténie de Glanzmann (anomalie quantitatve ou qualitative de la GPIIb-IIIa)


